
Intersexo
http://dcs.uqroo.mx/paginas/atlaspediatria/atlas022.html
 |
| Juguetes asexuados. Al fabricar muñecos que muestren anatómicamente los genitales masculinos y femeninos contribuirían a la larga y de alguna manera a evitar desde la más tierna infancia los engañitos con respecto a la sexualidad que desafortunadamente por considerarse tabú distorsionan las relaciones amoroso-genito-sexuales del adulto. |
Hace algunas décadas había escasos recursos para establecer la diferenciación sexual. Existían casos de pacientes con hipospadias severos a quienes era necesario identificarles el sexo posteriormente, pues a pesar de un reconocimiento propedéutico meticuloso, de estudios de uretrocistografía, de la determinación de los 17 cetoesteroides y de la laparotomía pélvica la duda empañaba la conducta por seguir.
Con base en los datos clínicos, después de un cuidadoso escrutinio, en ocasiones no se sabe si lo que se observa es un pene chico o un clítoris grande; labios mayores escrotalizados o escroto bífido, seno urogenital o hipospadias escrotoperineal y, más aún, si lo que se palpa es una punta de hernia, un testículo rudimentario o quizás un ovario. No sería raro que los padres, de acuerdo con el predominio somático de dichos órganos, hayan calificado a sus hijos como varones siendo niñas y viceversa; incluso, los médicos que practicaban la laparotomía pélvica no podían reconocer con certeza si lo que se les presentaba era un testículo atrófico o un ovario quístico, o bien, si lo que se introducía por el anillo inguinal era el ligamento redondo o los elementos espermáticos rudimentarios o, más aún, en los que parecían varones determinar si existían o no las vesículas seminales. También acontecía que durante la laparotomía se encontraban órganos con apariencia de testículo, por un lado, y de ovario, por el otro, lo que acrecentaba la duda.
Ante esta problemática se recurría a la biopsia transquirúrgica, que al cabo de unos cuantos minutos informaba de testículo hipoplásico, en una muestra, y ovario hipoplásico, en la otra; así, se concluía que trataba de un hermafroditismo verdadero con base en la diferenciación gonadal histológica. En aquellos años se dejaba pasar el tiempo para contar con el elemento psicológico de diferenciación sexual, y posteriormente, se intentaba algún tratamiento correctivo.
A partir de 1953 se ahondó en el problema y aparecieron autores como Barr, Jost, Davidson, Brigg y Kuperman, entre otros, quienes demostraron que la constitución genética masculina depende de las secreciones de los testículos embrionarios y comprobaron que, si el componente masculino disminuye, el componente normal femenino progresa en una dirección genética de este sexo; con base en ello, enunciaron el dimorfismo sexual de las células somáticas y percibieron masas características de cromatina hipercromática en las biopsias de piel, mucosa bucal, sangre y líquido aminiótico; además, perfeccionaron las pruebas del sexo en casos anómalos. Los diversos trabajos convergieron integralmente, se apoyaron y dieron a la valoración terapéutica de estas condiciones de diferenciación sexual ambigua, una ayuda inconmensurable. Desde entonces, los estudios sistemáticos de estos niños se modificaron e hicieron posible que muchos de ellos se trataran quirúrgicamente cada vez a más temprana edad, con lo que se les restituía precozmente al sexo correspondiente.
La primera etapa de diferenciación sexual ocurre desde el momento de la fertilización, cuando el óvulo acepta al espermatozoide maduro. El óvulo ostenta un cromosoma X mientras que el espermatozoide aporta un genosoma X o Y. La fertilización de un óvulo con cromosoma X, que recibe un gameto masculino con cromosoma X, origina a una mujer (XX), mientras que la fertilización de un óvulo por una célula espermática que contenga un cromosoma Y, produce un varón (XY).
A la quinta o sexta semanas de gestación el sistema reproductivo fetal se ve influido por el factor genético sexual descrito. Durante ese tiempo, el embrión se desarrolla bisexualmente; los genitales internos primitivos para ambos sexos crecen. Si el feto está destinado, por la combinación cromosómica, a ser un varón, el componente medular de la gónada sufre la diferenciación histológica hacia la formación del tejido testicular, y a la inversa, si el feto está destinado a ser mujer, el componente cortical de la gónada primitiva, una o dos semanas más tarde, se diferenciará sexualmente en este sentido.
En el tiempo transcurrido hasta entonces, alrededor de ocho semanas de vida fetal, se desarrollan simultáneamente los sistemas reproductivos internos del hombre y de la mujer, llamados gonadoductos (el conducto de Wolff, en el sexo masculino, y el conducto de Müller, en el femenino), y a través de transformaciones propias originarán los elementos característicos de cada sexo. Tal vez debido a influencias genéticas, hormonales o enzimáticas, los aparatos reproductivos internos del sexo opuesto inducidos a la involución degeneran y permiten que los del sexo correspondiente se desarrollen de modo normal.
Los genitales externos se desarrollan a partir de un mismo origen, el tubérculo genital, que mantiene su potencialidad bisexual hasta entonces. Dicha yema bipotencial permitirá, por elongación, que el pene se integre y se forme la uretra peneana, el prepucio y el escroto, o bien, que la elongación sea mínima, para producir al clítoris, a los labios menores y a los labios mayores.
De esta secuencia en la determinación sexual depende una serie de errores que originan procesos patológicos y que complican la diferenciación sexual, pero a la determinación del sexo contribuirá el estudio de los corpúsculos cromáticos de Barr. La presencia de ellos hablará a favor de un sexo cromático positivo (femenino) y su ausencia indicará un sexo cromático negativo (masculino). El corpúsculo está presente en el 50 a 70% de las células del sexo femenino y en menos de 10% en las del sexo masculino.
El término de hermafroditismo se ha utilizado desde épocas remotas para designar a la ambigüedad del sexo y continúa vigente hasta la fecha, a tal grado que la mayoría de los autores reconocen las grandes divisiones del intersexo en:
- Seudohermafroditismo masculino.
- Seudohermafroditismo femenino.
- Hermafroditismo verdadero.
- Síndromes de disgenesia gonadal.
El seudohermafroditismo masculino es el tipo que se observa más comúnmente en los grados mayores de hipospadias. El hipospadias escrotoperineal o perineal de tipo vulviforme es el que más se presta confusión. Los hallazgos inmediatos más llamativos son un glande grueso, un falo corto o ausente, falta de meato uretral y una semejanza con el capuchón clitorídeo. El escroto es bífido, por lo que asemeja labios mayores escrotalizados. La uretra se abre al periné entre dos bordes membranosos delgados que simulan al introito vaginal. Los testículos pueden estar en las bolsas, en las ingles o en la cavidad adbominal.
El frotis bucal de Moore y Barr, la biopsia de piel, el cariotipo y la laparotomía con biopsia gonadal aclararán un alto porcentaje de las dudas. Si la cromatina resulta negativa, es casi seguro que el paciente sea masculino.
Desde hace años se han preconizado los estudios mencionados en la sección anterior para el recién nacido y ha sido posible en los primeros días hacer las correcciones anatómicas pertinentes durante la misma laparotomía, dentro de las que han destacado la orquidopexia inguinal sin cierre herniario. Posteriormente, a los dos años ya se ha resecado la cuerda uretral, se ha bajado más el testículo o los testículos y se ha procedido al cierre de los orificios herniarios. La conducta posterior es similar a la de los demás casos de hipospadias.
Con esta conducta se ha evitado el registro equivocado de los niños y se ha logrado conducirlos dentro del patrón sexual que les corresponden en su vestir, jugar y arreglo personal, factores que reafirmarán la diferenciación sexual.
Seudohermafroditismo femenino
El seudohermafroditismo femenino se produce por la hiperplasia adrenal congénita. La mayoría de las pacientes exhibe la forma genital pura y la minoría la variedad de las perdedoras de sodio (síndrome de Debre-Fibiger). La forma genital se caracteriza por el desarrollo exagerado del clítoris, que se manifiesta desde una simple hipertrofia, en un principio, para transformarse en órgano parecido a un pene acodado, posteriormente el capuchón del clítoris es exuberante y los labios mayores hipertróficos forman rodetes que estrechan la hendidura vulvar. Al separar estas últimas formaciones se aprecia una cavidad en donde desembocan la uretra y la vagina, o bien, un sistema genital externo cerrado; la vagina parece desembocar más arriba que lo habitual.
La hiperplasia adrenal congénita se debe a un defecto biosintético del cortisol en el cual la glándula suprarrenal no produce suficiente cortisol, la pituitaria aumenta la producción de corticotropina para estimular la corteza adrenal e iniciar un proceso hiperplásico y la capa interior de la zona reticular produce andrógenos. Este aumento de andrógenos tiende a producir virilidad en la mujer. Si hay producción de cortisol en forma incompleta las niñas no perderán sodio, pero si la falla en la producción de cortisol es total, además del cuadro genital podrán existir vómito, diarrea, anorexia, palidez y deshidratación, que pueden conducir a la muerte. La disminución del sodio y del cloro sanguíneos, la elevación de la reserva alcalina y el aumento del potasio son, a su vez, capaces de producir otras fallas hormonales que acentúan los rasgos genitales.
En el seudohermafroditismo femenino es común el aumento pondoestatural, que llega a sobrepasar el 50% del correspondiente a la edad de un niño normal.
El estudio imagenológico del sistema óseo denota la aceleración en el desarrollo de los núcleos epifisiarios, que ocasiona aumento de talla antes de los 11 años, a partir de los cuales el crecimiento se estanca si no se instituye el tratamiento adecuado. El hirsutismo púbico y axilar aparece progresivamente y puede abarcar la región del labio y la barba. Los senos, la menstruación y los caracteres sexuales secundarios femeninos faltan habitualmente si no se establece el tratamiento oportuno y adecuado.
Ante todas las sospechas clínicas enunciadas, la investigación de la cromatina positiva, la elevación de los 17 cetosteroides neutros del pregnandiol y la eliminación considerable del pregnantriol, serán bases más que suficientes para ratificar el diagnóstico.
Otras causas de seudohermafroditismo femenino sin hiperplasia adrenal, las constituyen los casos en que existen antecedentes de administración de progesterona o andrógenos en las madres grávidas; cuando las madres son portadoras de arrenoblastoma, y los casos de niñas a quienes durante largo tiempo se les ha administrado andrógenos desde el nacimiento. Asimismo, se ha informado de casos de seudohermafroditismo femenino sin causa hormonal ostensible y casos de tumores virilizantes de la glándula suprarrenal, en los que estudios radiológicos especiales, son capaces de demostrar el tumor.
El tratamiento de estos enfermos es medicoquirúrgico. El primero consiste en administrar cortisona o hidrocortisona, en dosis adecuadas a la edad, iniciadas por vía intramuscular y mantenidas por vía oral. Cuanto más precozmente se inicie el tratamiento menos complicaciones secundarias se presentarán en estas niñas, aunque tienda a persistir la hipertrofia clitorídea. En las pacientes perdedoras de sodio, el tratamiento aconsejable es a base de desoxicorticosterona (DOCA) y adición de sal.
Sobre el manejo quirúrgico del seudohermafroditismo femenino, apoyan la corrección de la vagina de situación baja y la de situación alta, a través de una operación transperineal por la cual se pueda colocar abajo, en el perineo. Se cree que la corrección normalizadora del perineo debe efectuarse tan precozmente como sea posible e, incluso, antes de iniciar la terapia esteroidea. Unos autores abogan por la extirpación sistemática del clítoris y otros opinan que esto se debe evaluar después de la terapia esteroidea.
Hermafroditismo verdadero
El hermafroditismo verdadero puede presentar las características clínicas del sexo ambiguo y ser un problema de diferenciación sexual. La laparotomía propedéutica y la biopsia gonadal transquirúrgica rubricarán este tipo de anomalía. Con base en estos últimos estudios se pueden clasificar tres tipos anatómicos: a) los que tienen un testículo de un lado y un ovario del otro (alternante); b) los que tienen dos ovarios y dos testículos (bilateral), y c) los que en un lado tienen ovario y testículo o una gónada diferenciada (unilateral).
En estos enfermos no se sugiere, a pesar de contar con un patrón definido de cromatina sexual, llevar a cabo algún procedimiento plástico, sino hasta que el niño llegue a la edad de la pubertad. El patrón cromosómico y la clínica le brindarán al paciente anticipadamente un sexo para que se desenvuelva acorde con él, pero se cree prudente no intentar la intervención quirúrgica sino hasta que exista una tendencia declarada por alguno de los sexos, para evitar muchos errores irremediables. Si el joven adquiere los hábitos y deseos del sexo masculino se intentará retirar lo anatómicamente femenino y mejorar lo masculino, incluyendo el tratamiento hormonal apropiado; si el sujeto anhela la femineidad se formará la vagina y se le extirparán los testículos.
Síndromes de disgenesia gonadal
Los síndromes de disgenesia gonadal más comunes son los de indiferenciación gonádica de Turner y el síndrome de Klinefelter.
Síndrome de Turner.
Éste ocurre en mujeres; el cariotipo es de 45 cromosomas en lugar de 46 y su anomalía se caracteriza por la presencia de cromosomas sexuales XO. Las personas son de aspecto femenino con órganos genitales femeninos infantiles y de talla corta; el útero está presente pero los ovarios son aplásicos. Este síndrome suele ir acompañado de otras anomalías como son: cuello alado o membranoso, alteraciones cardiovasculares, problemas oculares y articulares y sordera, que puede contribuir a cierto grado de retraso mental. La hormona foliculoestimulante hipofisaria está elevada y la carencia de los estrógenos se puede comprobar por medio de frotis vaginal.
El cirujano colabora con los internistas en la laparotomía biópsica, en la extirpación de las gónadas virilizantes y en la corrección de las anomalías coexistentes.
Síndrome de Klinefelter.
Éste es un trastorno que ocurre en el varón. Las personas tienen apariencia masculina y caracteres sexuales secundarios masculinos, pero con testículos muy pequeños y azoospérmicos por gran atrofia testicular. En este síndrome existen 47 cromosomas (XXY) en lugar de 46 y la prueba cromatínica de Barr es generalmente positiva. La hormona foliculoestimulante hipofisaria está elevada, pero los estrógenos permanecen normales. La biopsia del testículo revela la atrofia de los conductos seminíferos.
El cirujano colabora con los internistas para la biopsia, para la extirpación de las glándulas o para la exéresis de la ginecomastia.
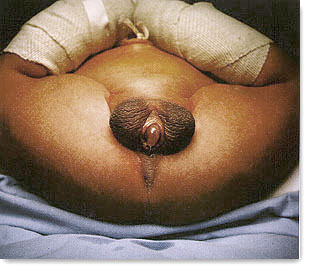 | Pseudo hermafroditismo masculino. Entre otras anomalías no existe el dartos prepucial. La laparotomía temprana, dirigida y correctiva iniciará el ajuste sexual orgánico en base también la cromatina sexual investigada por legrado de mucosa oral, folículos, etc. |
| Pseudohermafroditismo. Pene atrófico o clitoris hipertrófico. Hipospadias perineo-fálico o sinus vaginal. Grandes labios escrotalizados o escroto bífido. Pensar en disgenesia gonadal (síndrome de Turner y síndrome de Klinefelter). | 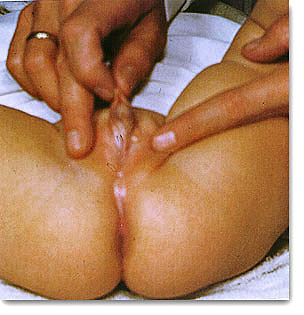 |
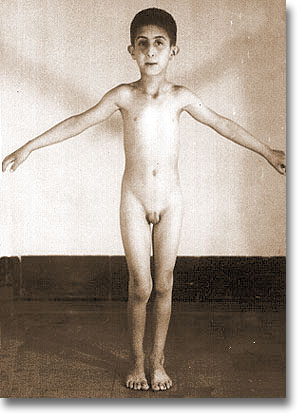 | Pseudohermafrotismo femenino. Apariencia feminoide de su cuerpo. |
| Pseudohermafroditismo con ausencia o hipoplasia del pene (grupo D). | 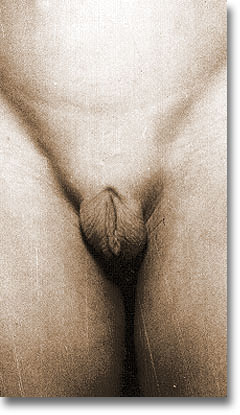 |
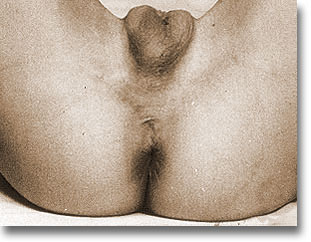 | Pseudohermafroditismo femenino. Bolsas escrotales ocupadas. Pene atrófico. |
| Pseudohermafroditismo. Coloboma prepucial de pene rudimentario o prepucio clitoroideo exhuberante. Laparoscopía o laparotomía precoz dirigida con previos estudios citocromosómicos hormonales, genética molecular, ultrasonido, cistouretrografía, estimulación gonadal mediante HCG o LH/FSH. Asignación sexual. | 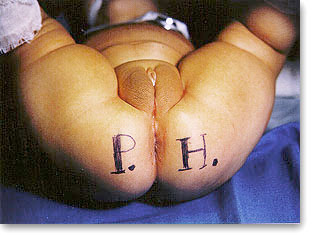 |
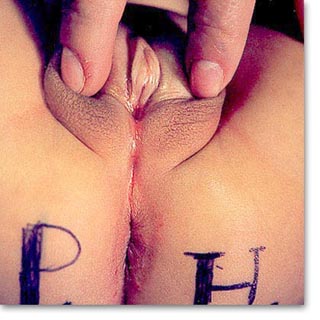 | Intersexo. Hermafroditismo verdadero. Puede presentar características clínicas de sexo ambiguo. Laparotomía prepedéutica, biopsia gonadal (un testículo de un lado y un ovario del otro, alternante; dos ovarios y dos testículos, bilateral; de lado ovario y testículo y del otro lado igual). |
| Hermafroditismo verdadero (b) ovarios y testis en ambos lados. Pene chico o clítoris grande. Patología poco común. Laparotomía exploradora y dirigida. Exéresis de lo conveniente previa biopsia, constitución genética, cromatina hipercromática, 17 cetosteroides y otros. |  |
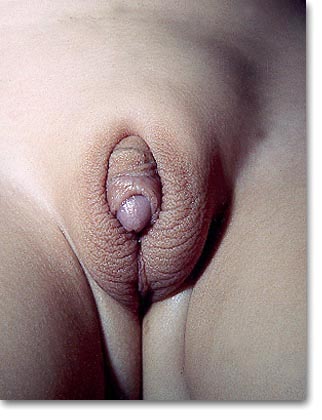 | Pseudohermafroditismo femenino. Marcada hipertrofia del clítoris que simula un pene con labios escrotalizados. Cromatina positiva, 17 cetoesteroides neutros, pregnandiol, pregnatriol. Tumores suprarrenales. |
| Sexo ambiguo. Se palpa lo que parecen ser testículos ectópicos suprapúbicos. Los estudios cromosómicos y endocrinológicos son coadyubantes. | 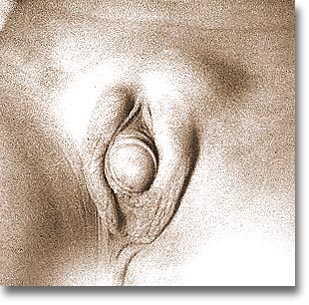 |
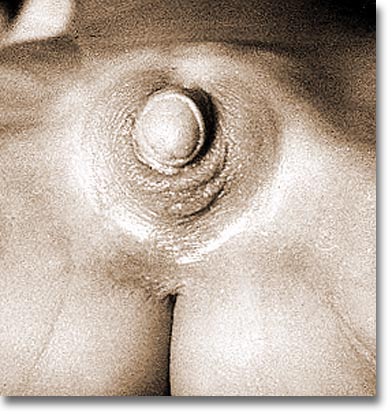 | Intersexo. En decúbito dorsal desaparece el abolsamiento escrotal y su posible contenido. |
| Los corpúsculos cromáticos de Moore y Barr opinan del sexo cromático positivo (femenino) y su ausencia de un sexo cromático negativo (masculino). Frotis bucal, biopsia de piel, cariotipo y laparotomía precoz aclararán en un alto porcentaje las dudas. El pseudohermafroditismo femenino se produce por la hiperplasia adrenal congénita. La mayoría la forma genital pura y la minoría la variedad de perdedoras de sodio (síndrome de Debre-Fibiger). | 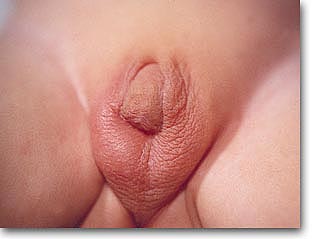 |
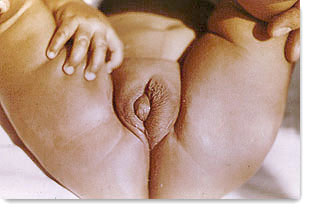 | Sexo ambiguo. Dos grados severos de hipospadias peneoescrotales se prestan a confusión, más aún la distopia del meato uretral. La clitoridectomía estará indicada previos y exhaustivos estudios. |
| Prepucio exhuberante. Pseudohermafroditismo femenino. Fusión labial y aumento de tamaño del clítoris. Hiperplasia adrenal congénita. Debre-Fibiger (perdedoras de sodio). Clitoridoplastia. | 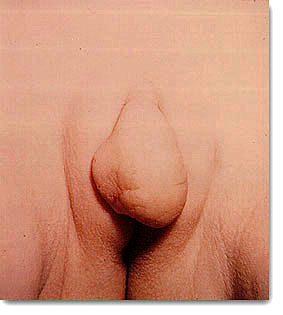 |
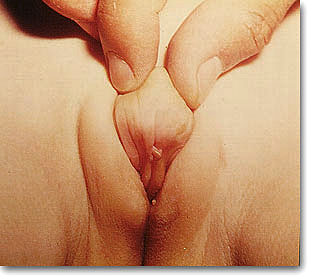 | En estos casos se impone la clitoridectomía para proporcionar una apariencia cercana a la normalidad. Tracción del clítoris, catéter uretral, incisión cóncava hacia arriba, sección del ligamento suspensorio, ligadura y corte de ambos cuerpos cavernosos y sutura vertical por planos. Abertura del sinus urogenital. |
No hay comentarios.:
Publicar un comentario